lentiviral Cas9ヌクレアーゼ試薬
生物学的に関連する細胞タイプを用いた確かなゲノム編集のための、レンチウイルスCRISPR-Cas9コンポーネント
Cas9ヌクレアーゼ安定発現細胞集団を作製するためのレンチウイルス粒子またはプラスミドDNAをご用意しています。大容量製品の価格についてはお問い合わせださい。
Try our new: Dharmacon™ Strict-R™ Inducible Cas9 Lentiviral System for dual controlled gene knockout in diverse cell types.
CRISPR-Cas9システムにおけるCas9ヌクレアーゼ
CRISPR関連酵素Cas9は、ゲノムDNAの標的認識とDNA二重鎖切断(double-strand breaks: DSB)のためにガイドRNAを必要とするRNA-guidedエンドヌクレアーゼです。
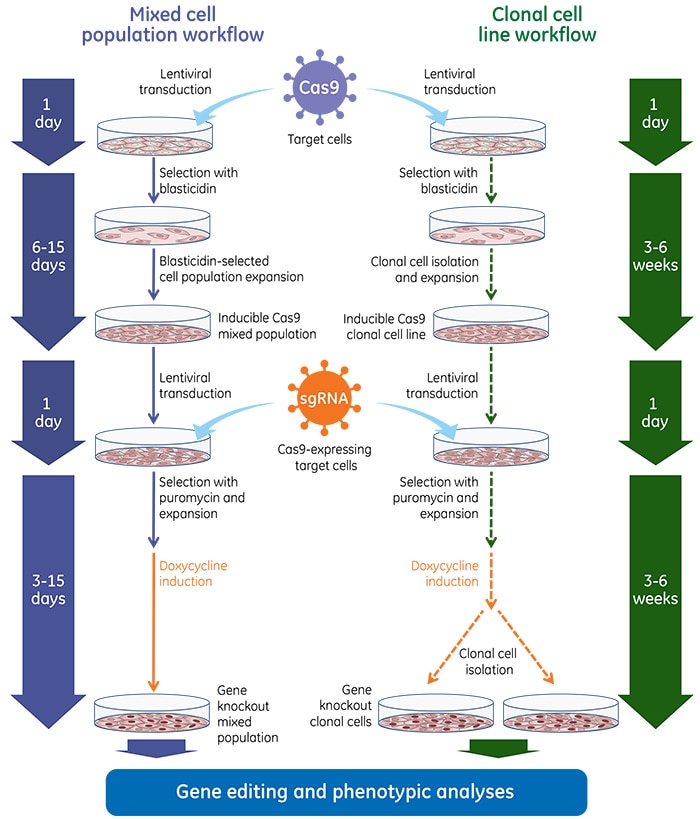
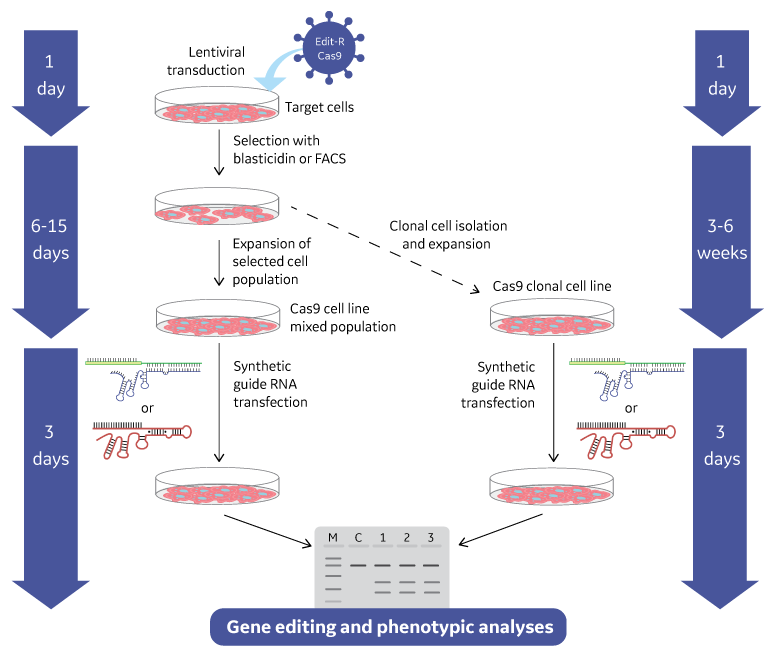
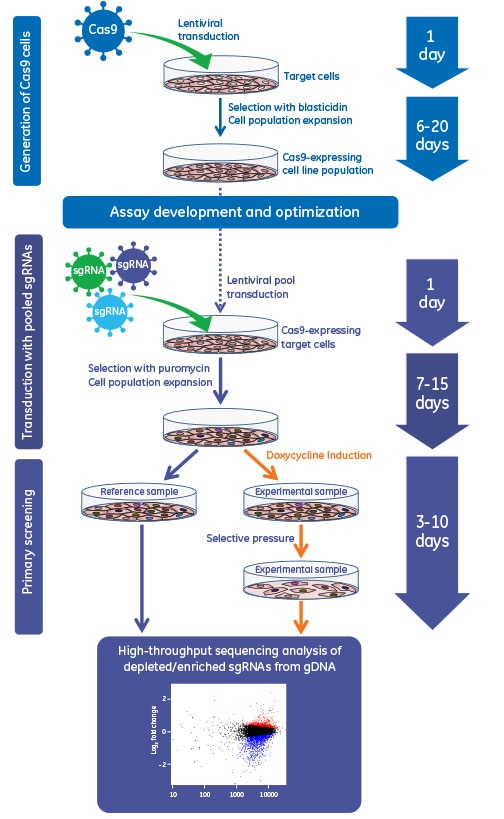
lentiviral Cas9試薬は、Cas9ヌクレアーゼを発現する細胞株の迅速な作製を容易にし、プール化lentiviral sgRNAスクリーニング、crRNAと共に使用するアレイ化スクリーニング、または単一遺伝子および/または複数の遺伝子を標的とする複数のガイドRNAの評価など、多くのゲノム編集アプリケーションに使用されています。
lentiviral Cas9ヌクレアーゼ発現ベクターには、S. pyogenes Cas9ヌクレアーゼのヒトコドン最適化バージョンを、さまざまな構成的または誘導的プロモーターの制御下が含まれています。全てのフォーマットは、精製されたレンチウイルス粒子またはプラスミドDNAとしてご提供しています。
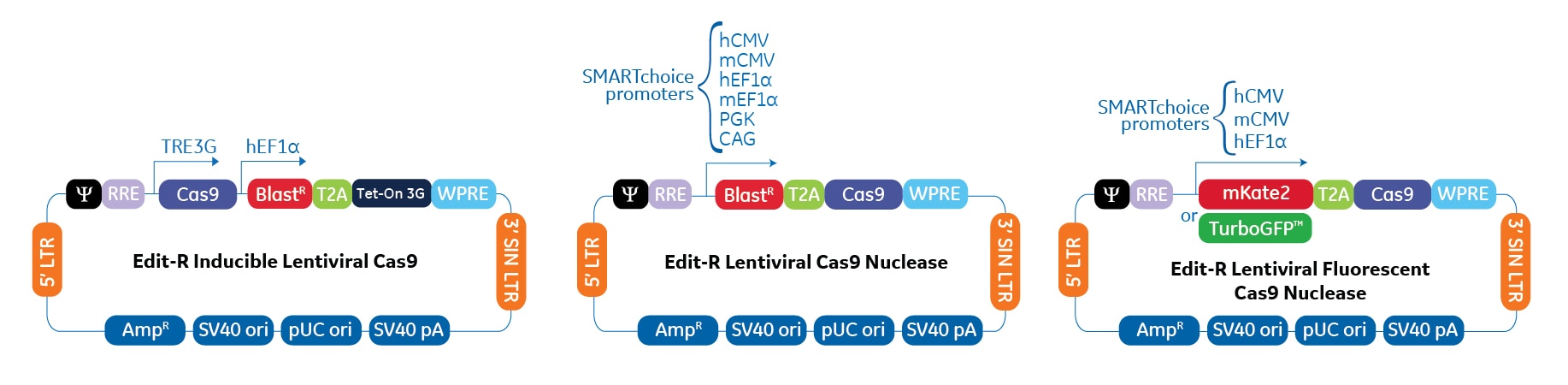
lentiviral Cas9 expression vector highlights
- ブラストサイジン耐性マーカー(BlastR)または蛍光マーカー(mKate2 または TurboGFP™)をを用いた選択が可能です。
- 即時形質導入が可能な濃縮精製レンチウイルス粒子であり、最小1x107 TU/mLの機能力価です。
- お客様独自のレンチウイルス粒子を生産するためのパッケージング細胞株への直接トランスフェクション用に、認証されたエンドトキシンフリープラスミドDNAとしてもご利用いただけます。
- 6 つのSMARTchoice構成的プロモーターのいずれかを使用してコンストラクトをカスタマイズし、目的の細胞株でのCas9の発現を最適化できます。
- Cas9の発現を一時的に制御する必要がある場合、またはバックグラウンド発現を最小限に抑えた安定した細胞株を作製する場合は、誘導型Cas9ベクターの厳密な制御をご利用いただけます。
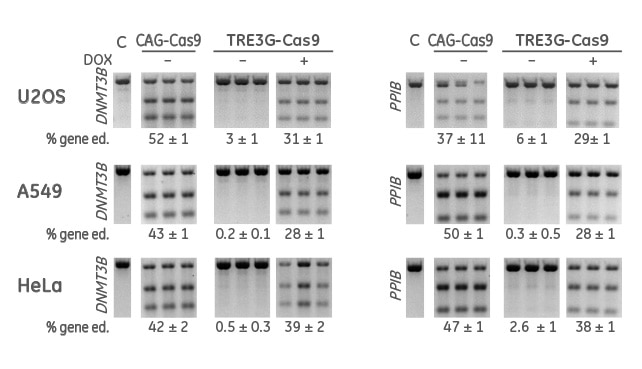
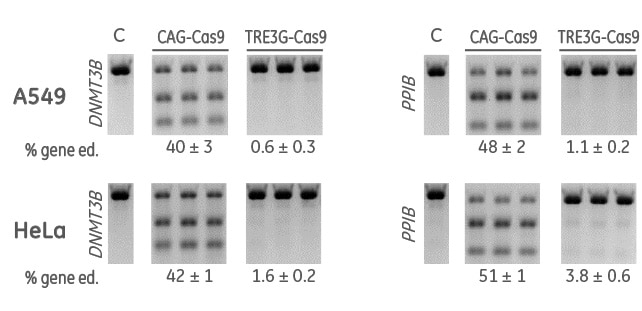
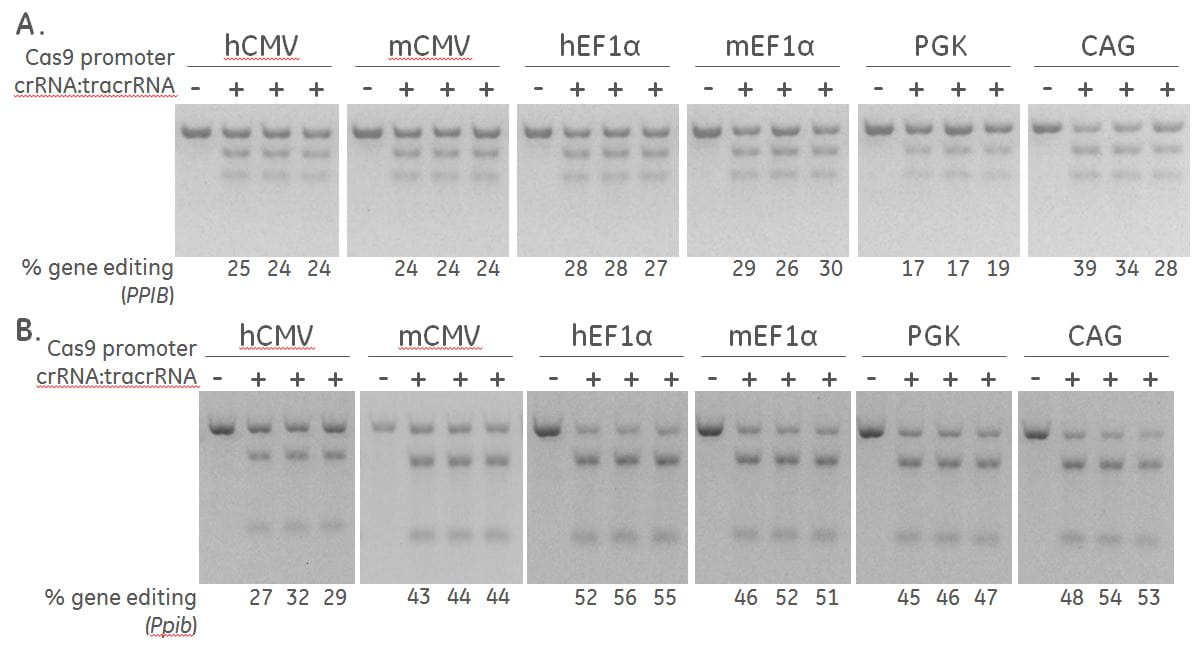
すべてのRNA pol IIプロモーターが異なる細胞環境で同等にアクティブではない
Cas9ヌクレアーゼの転写を制御する任意のプロモーターの活性は、生物系によって大きく異なる可能性があり、Cas9の発現レベルが変化し、ひいてはDNA編集のレベルが変化します。したがって、細胞株または細胞タイプに最適なプロモーターを選択することは、実験におけるゲノム編集の程度に影響します。
Cas9ヌクレアーゼを発現するためのSMARTchoiceプロモーターオプション| Promoter | Description |
|---|---|
| hCMV | human cytomegalovirus immediate early promoter |
| mCMV | mouse cytomegalovirus immediate early promoter |
| hEF1α | human elongation factor 1 alpha promoter |
| mEF1α | mouse elongation factor 1 alpha promoter |
| PGK | mouse phosphoglycerate kinase promoter |
| CAG | chicken beta actin hybrid promoter |
| TRE3G | doxycycline-inducible promoter |