CRISPRmod(CRISPRiおよびCRISPRa)実験では、効率的な転写調節のために、細胞内で2つのコンポーネントをデリバリーおよび/または発現させる必要があります。1つは、遺伝子を標的とするガイドRNAであり、もう1つは転写リプレッサー(SALL1およびSDS3)またはアクチベーター(VPR)のいずれかに融合したヌクレアーゼ活性を不活性化したCas9(dCas9)です。いずれかの成分の量が不十分な場合、標的遺伝子の転写抑制または転写活性化が非効率的になるため、成功を確実にするために適切なコントロールを使用する必要があります。
コントロールを使用する目的:
- 化学合成ガイドRNAトランスフェクション条件の最適化
- レンチウイルスdCas9発現を駆動するための最適なプロモーターの決定
- dCas9 mRNAによる最適なコトランスフェクション条件の特定
- 実験の一貫性の確保、および考えられるバックグラウンド効果のコントロール
機能獲得および機能喪失実験のためのCRISPRModコントロール
最適な実験条件を確立し、継続的に成功する遺伝子転写抑制/活性化を確実にするために、特徴付けられた標的遺伝子に対してポジティブコントロールを使用することをお勧めします。
non-targeting(ネガティブ)コントロールは、注釈付きのヒトゲノムに相補的な標的配列がないガイドRNAであるため、このコントロールによって特定の遺伝子のベースライン発現は変化しないことが期待されます。CRISPRmod実験では、mRNAとタンパク質のレベルが時間とともに変化する可能性があります。発現レベルを検出する際には、これを考慮に入れる必要があります。qPCRとウエスタンブロッティングを使用して、未処理およびnon-targetingコントロールと比較したmRNAおよびタンパク質レベルの変化を決定し、遺伝子発現調節効果を観察します。Read more about CRISPRi applications.
CRISPRa controls
To establish optimal experimental conditions and ensure ongoing successful activation, it is recommended to use a species-specific CRISPRa positive control for a characterized gene target. The CRISPRa positive controls target human or mouse Titin (TTN) or POU class 5 homeobox 1 (POU5F1) genes. Read more about CRISPRa applications.
CRISPRi化学合成sgRNAポジティブコントロールの確実な遺伝子ノックダウン

dCas9-SALL1-SDS3 mRNAと化学合成CRISPRi sgRNAポジティブコントロールの共導入による頑健な遺伝子転写抑制。K562およびJurkat細胞に、dCas9-KRABまたはdCas9-SALL1-SDS3 mRNA(2 µg)、およびPPIBとSEL1Lを標的とするプールされたCRISPRi化学合成sgRNA(5 µM)をLonza 96-well Shuttle systemを介してヌクレオフェクションしました。WTC-11 hiPS細胞を、Lonza社の96ウェルシャトルシステムを用いて、dCas9-KRABまたはdCas9-SALL1-SDS3 mRNA(1 μg)と、PPPIBまたはSEL1Lを標的とするプールされたCRISPRi化学合成sgRNA(3 μM)でヌクレオフェクションし、ヌクレオフェクションの 72 時間後に細胞を回収しました。全RNAを単離し、RT-qPCRを用いて相対的遺伝子発現を測定しました。各標的遺伝子の相対遺伝子発現は、ハウスキーピング遺伝子としてGAPDHを使用した∆∆Cq法で行い、non-targetingコントロールに対して正規化しました。
dCas9-VPR安定発現細胞における化学合成CRISPRa crRNA:tracrRNAによる効率的な遺伝子の転写活性化

dCas9-VPR安定発現細胞における化学合成CRISPRa crRNA:tracrRNAによる効率的な遺伝子の転写活性化。dCas9-VPRを安定発現するように改変されたHEK293T、U2OS、MCF 10A、NIH/3T3を10,000細胞/ウェルでプレーティングし、DharmaFECTトランスフェクション試薬を使用して、POU5F1およびTTNを標的とする化学CRISPRa crRNA:racrRNA(25 nM)をトランスフェクトしました。K562細胞には、POU5F1およびTTNを標的とする化学合成CRISPRa crRNA:tracrRNA(400 nM)をエレクトロポレーションしました。トランスフェクションの72時間後に細胞を回収し、RT-qPCRを使用して相対的な遺伝子発現を測定しました。各遺伝子の転写活性化倍率は、ハウスキーピング遺伝子としてGAPDHを使用するCq法で計算し、non-targetingコントロールに対して正規化しました。
dCas9-VPR安定発現細胞株におけるレンチウイルスCRISPRa sgRNAによる効率的な遺伝子転写活性化
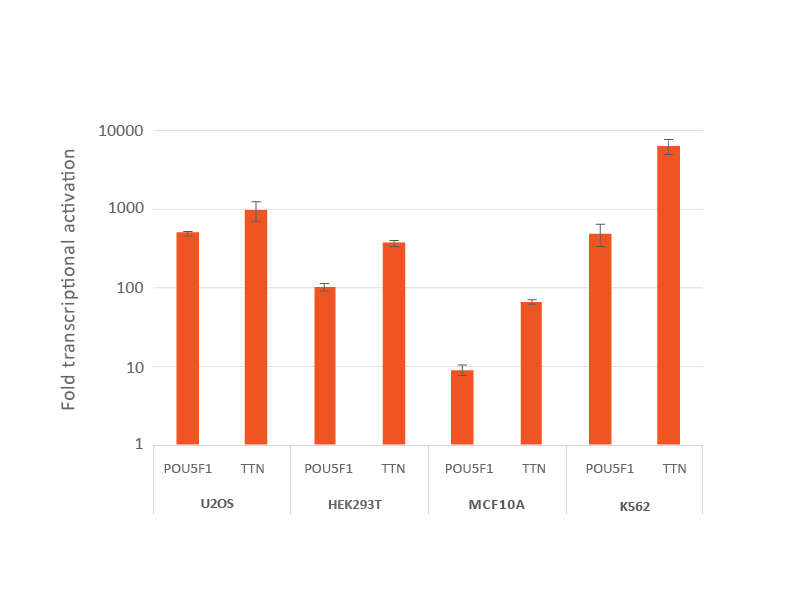
dCas9-VPRを安定発現するように改変されたU2OS、HEK293T、MCF 10A、およびK562を10,000細胞/ウェルでプレーティングし、POU5F1またはTTNを標的とするCRISPRa sgRNAレンチウイルス粒子をMOI 0.3で形質導入して、単一の改変細胞を得ました。細胞を2 µg/mLピューロマイシンで4日間選択した後、RT-qPCRで分析しました。各遺伝子の相対的発現は、ハウスキーピング遺伝子としてGAPDHを使用するΔΔCq法で計算し、non-targetingコントロールに対して正規化しました。
Order CRISPRiコントロール
CRISPRi synthetic sgRNA(化学合成sgRNA)ポジティブコントロール
プール化フォーマットまたは個別の化学合成sgRNAコントロールであり、遺伝子転写抑制の効率を最大化するための実験条件の検討、および最適化の確認のために使用します。
CRISPRi synthetic sgRNA(化学合成sgRNA)non-targetingコントロール
標的遺伝子特異的sgRNAの非存在下でCRISPRiコンポーネントに対するベースラインの細胞応答を評価します。
CRISPRi lentiviral sgRNAポジティブコントロール
十分に特徴付けられた遺伝子を標的とする検証済みのsgRNAをポジティブコントロールして用いることで、遺伝子転写抑制の効率を最大化するための実験条件の検討、および最適化の確認のために使用します。
CRISPRi lentiviral sgRNA non-targetingコントロール
標的遺伝子特異的sgRNAの非存在下でCRISPRiコンポーネントに対するベースラインの細胞応答を評価します。
Order CRISPRaコントロール
CRISPRa synthetic crRNA(化学合成crRNA)ポジティブコントロール
プールフォーマットまたは個別の化学合成crRNAコントロールであり、遺伝子転写活性化の効率を最大化するための実験条件の検討、および最適化の確認のために使用します。
CRISPRa synthetic crRNA(化学合成crRNA)non-targetingコントロール
標的遺伝子特異的crRNAの非存在下でCRISPRaコンポーネントに対するベースラインの細胞応答を評価します。
CRISPRa synthetic sgRNA positive controls
Pooled or individual sgRNA controls for assessment of optimal experimental conditions for gene activation
CRISPRa synthetic sgRNA non-targeting controls
Non-targeting controls to evaluate baseline cellular responses to CRISPRa components in the absence of gene target-specific sgRNA
CRISPRa lentiviral sgRNAポジティブコントロール
十分に特徴付けられた遺伝子を標的とする検証済みのsgRNAをポジティブコントロールして用いることで、遺伝子転写活性化の効率を最大化するための実験条件の検討、および最適化の確認のために使用します。
CRISPRa lentiviral sgRNA non-targetingコントロール
標的遺伝子特異的sgRNAの非存在下でCRISPRaコンポーネントに対するベースライン細胞応答を評価します。
Order all-in-oneコントロール
CRISPRi all-in-one lentiviral sgRNAポジティブコントロール
十分に特徴付けられた遺伝子を標的とするall-in-oneのレンチウイルスsgRNAをポジティブコントロールして用いることで、遺伝子転写抑制の効率を最大化するための実験条件の検討、および最適化の確認のために使用します。
CRISPRi all-in-one lentiviral sgRNA non-targetingコントロール
バイオインフォマティクスに基づいて設計されたall-in-one non-targeting sgRNAコントロールは、標的遺伝子特異的sgRNAの非存在下でCRISPRiコンポーネントに対するベースラインの細胞応答を評価します。
CRISPRa all-in-one lentiviral sgRNAポジティブコントロール
十分に特徴付けられた遺伝子を標的とするall-in-oneのレンチウイルスsgRNAをポジティブコントロールして用いることで、遺伝子転写活性化の効率を最大化するための実験条件の検討、および最適化の確認のために使用します。
CRISPRa all-in-one lentiviral sgRNA non-targetingコントロール
バイオインフォマティクスに基づいて設計されたall-in-one non-targeting sgRNAコントロールで、標的遺伝子特異的sgRNAの非存在下でCRISPRaコンポーネントに対するベースラインの細胞応答を評価します。