特定のゲノム位置に二本鎖切断(double-strand breaks: DSB)を作るためのガイドRNA(gRNA)の設計は簡単なよう(単にPAM部位を探すだけ)に見えますが、オフターゲット効果を避けながら、ゲノム編集が目的の遺伝子座で確実に行われるようにするには、考慮すべき多くの要素があります。
我々は、ゲノム編集を成功させるために、機能性と特異性を念頭に置いてEdit-Rアルゴリズムを開発しました。gRNAが特異的でなかったり機能的でなかったりすると、望ましくない遺伝子発現や望ましくない細胞効果を引き起こす可能性があります。特異性とは、gRNAがどれだけ特定の遺伝子を標的とできるかを意味し、機能性とは、gRNAが効果的に遺伝子をオンまたはオフにする能力を意味します。
特異性評価を含むEdit-Rアルゴリズムは、各gRNAのスコア付けに使用され、機能的タンパク質ノックアウトの可能性が最大で、オフターゲット編集が最小となるよう予めデザインされたgRNAを提供します。これらの試薬は、アレイ化スクリーニング用の化学合成および化学修飾RNAとして、またはプールスクリーニング用の発現レンチウイルス製品としてご利用いただけます。
効率的な遺伝子機能研究のためのCRISPRノックアウト・アルゴリズム
CRISPR-Cas9は、遺伝子機能を調べるために機能的遺伝子ノックアウトを引き起こす非常に効果的なツールですが、初期の研究では、ガイドRNAの機能性に大きなばらつきがあることが示されました。高性能のガイドRNA配列は、ゲノムDNAを切断すると同時に、挿入や欠失(indel)を形成して遺伝子を効果的に破壊し、改変されたmRNAから機能的タンパク質への翻訳を阻害する必要があります。機能的遺伝子ノックアウトを生み出す可能性が最も高いgRNAを設計するために、我々は機能的遺伝子破壊を用いたデータで訓練されたアルゴリズムを開発しました。
機能的遺伝子ノックアウトによる表現型データ
この目的のために、プロテアソーム機能に関与する複数の遺伝子ターゲットに対する幅広い種類のcrRNAを、組換えU2OSユビキチン-EGFPプロテアソーム細胞株でテストしました。この細胞株は、EGFPタンパク質の迅速な分解を可能にし、EGFPのベースライン蛍光を低くする、切断不可能な変異型ヒトユビキチン(Gly76Val)と融合したEGFPレポーターを安定に発現しています。低分子阻害剤、遺伝子阻害、遺伝子ノックアウトなどによるプロテアソーム関連成分の破壊は、EGFP分解を阻害し、蛍光シグナルを増加させます(Figure 1)。
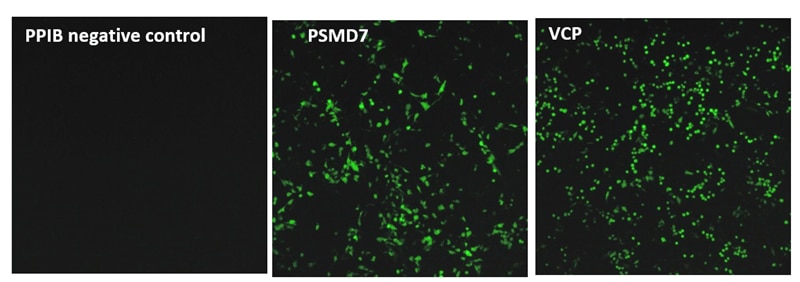 Figure 1. 組換えU2OSユビキチン-EGFPプロテアソーム細胞株(Ubi[G76V]-EGFP)を、hCMVプロモーターによって駆動されるCas9とblasticidin耐性遺伝子を含むレンチウイルス粒子で安定的に形質導入しました。安定的に導入された細胞集団をブラストサイジンで選択し、プロテアソーム経路の遺伝子を標的とするcrRNA:tracrRNAによるその後のトランスフェクションに使用しました。トランスフェクションのために、Ubi[G76V]-EGFP-Cas9細胞を、底が透明な黒色96ウェル組織培養プレートに4,300細胞/ウェルでプレーティングしました。翌日、DharmaFECT 4 Transfection Reagent(0.07 µg/well)を用いて、PPIB、PSMD7またはVCP遺伝子を標的とする50 nMの化学合成crRNA:tracrRNAでトランスフェクトしました。72時間後、Envisionプレートリーダーを用いてEGFP蛍光を測定しました。
Figure 1. 組換えU2OSユビキチン-EGFPプロテアソーム細胞株(Ubi[G76V]-EGFP)を、hCMVプロモーターによって駆動されるCas9とblasticidin耐性遺伝子を含むレンチウイルス粒子で安定的に形質導入しました。安定的に導入された細胞集団をブラストサイジンで選択し、プロテアソーム経路の遺伝子を標的とするcrRNA:tracrRNAによるその後のトランスフェクションに使用しました。トランスフェクションのために、Ubi[G76V]-EGFP-Cas9細胞を、底が透明な黒色96ウェル組織培養プレートに4,300細胞/ウェルでプレーティングしました。翌日、DharmaFECT 4 Transfection Reagent(0.07 µg/well)を用いて、PPIB、PSMD7またはVCP遺伝子を標的とする50 nMの化学合成crRNA:tracrRNAでトランスフェクトしました。72時間後、Envisionプレートリーダーを用いてEGFP蛍光を測定しました。
我々は、プロテアソーム機能に必須であると以前に同定された10個の遺伝子のコード領域をターゲットとする全てのgRNA(NGG PAMの上流)を合成し、1,100以上のデータポイントを作成しました。gRNAは異なるレベルのEGFP蛍光を示し、機能的遺伝子破壊のレベルが異なることを示しました。すなわち、我々はこのデータを機械学習に利用し、遺伝子ノックアウトの成功と相関するデザイン特性を同定しました。機能的な遺伝子破壊は、遺伝子の位置と標的配列の構成の両方に依存します。Figure 2はVCP(A)またはPSMD8(B)遺伝子を標的にして得られたデータの例です。VCP遺伝子は大きく、グラフ中で色分けされている17個のエクソンから構成されています。この遺伝子の全長を標的とするためには266個の化学合成crRNAが必要でした。開始点からの距離をEGFPシグナルの強度に対してプロットしたもので、EGFPシグナルの増加はVCP遺伝子とタンパク質の機能的ノックアウトを示しています。機能性の高いcrRNAは、初期あるいは後期エクソンを含む異なるエクソンにわたって同定されました。例えば、1つの初期エクソン(エクソン2)には、高機能なcrRNAが1つ存在し、ほとんどが低機能なcrRNAでした。エクソン14では、高機能なcrRNAに隣接して、高機能なcrRNAが多数存在しますが、非機能なcrRNAや低機能なcrRNAも存在する領域が見られました。これは明らかに、配列特異的な因子がgRNAの機能性に寄与していることを示しています。PSMD8遺伝子(B)は7つのエクソンから構成されています。興味深いことに、最初のエクソン(エクソン1)の最初の200塩基を標的とするgRNAは、機能的な遺伝子破壊(EGFPシグナルの増加なし)には至りませんでしたが、一方で高い indel形成能を示しました(データ非掲載)。我々は、この遺伝子が代替転写開始点(transcriptional start site: TSS)を利用している可能性が高いことを発見し、初期エクソンでgRNAを選択することがいかに機能的遺伝子ノックアウトにつながらないかを明確に示しました。FANTOMデータベースの約15%の遺伝子は、タンパク質の合成において初期エクソンのスキップにつながる可能性のある代替TSSを有しており、下流の機能的タンパク質解析のためのgRNA設計においてこれを考慮する必要があります。
機械学習を用いて効率的なgRNAの特徴を解析し、sgRNAの活性を予測する特徴を見出しました。その特徴には、配列の特徴(ヌクレオチド位置、ジヌクレオチド組成、GC含量、DNA-RNA複合体のTm、PAM配列、オーバーラップPAMなど)、標的遺伝子の特徴(遺伝子内の距離、開始可能部位の数、開始・停止コドンからの距離、イントロン/エクソン接合部からの距離、+/-鎖を標的とする、など)が含まれます。gRNAの活性予測を最適化するために、これらの特徴に基づいた定量的モデルを開発し、このモデルをsgRNAの設計に使用するツールを作成しました。設計アルゴリズムの性能は、ROC(Receiver Operating Characteristic)曲線を作成することでテストしました(Figure 3)。我々のモデルの平均曲線下面積(AUC)値は 0.78 であり、 高い予測力とこのアルゴリズムで特徴付けられたガイドRNAに対する高い信頼性を示しています。
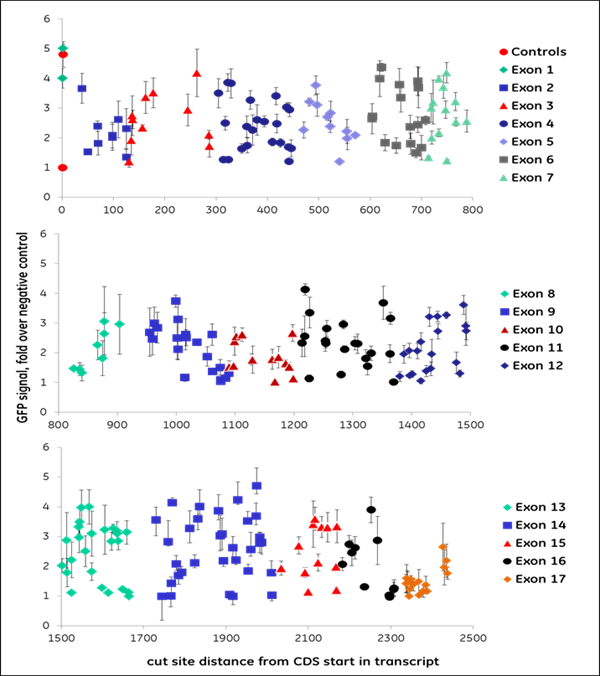
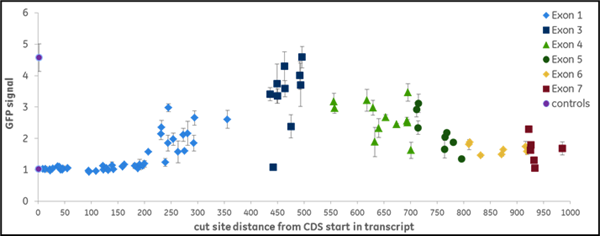
Figure 2. 遺伝子機能破壊を引き起こすgRNAの能力は様々です。組換えU2OS Ubi[G76V]-EGFP -Cas9細胞に、(A)VCP遺伝子または(B)PSMD8遺伝子のコード領域の長さに沿って化学合成crRNA:tracrRNA複合体をトランスフェクトしました。Envisionプレートリーダーを用いて、トランスフェクション後72時間でEGFP蛍光を測定しました。
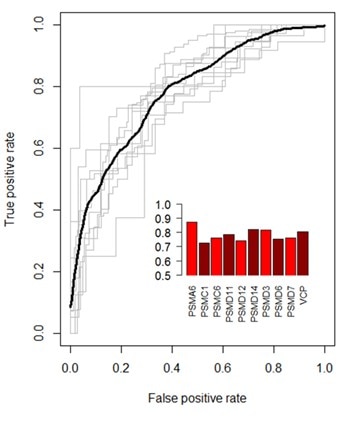 Figure 3. CRISPR gRNA予測アルゴリズムの性能検証。ROC曲線は真陽性の割合と偽陽性の割合を比較し、分類モデルの性能の指標となります。モデルは9つの遺伝子のすべての可能な組み合わせで学習され、残りの保留遺伝子で個別にテストされました。各灰色線は、保留遺伝子に対するROC曲線を示しています。黒線は平均ROC曲線。棒グラフの挿入は、各遺伝子の曲線下面積(AUC)を示しています。
Figure 3. CRISPR gRNA予測アルゴリズムの性能検証。ROC曲線は真陽性の割合と偽陽性の割合を比較し、分類モデルの性能の指標となります。モデルは9つの遺伝子のすべての可能な組み合わせで学習され、残りの保留遺伝子で個別にテストされました。各灰色線は、保留遺伝子に対するROC曲線を示しています。黒線は平均ROC曲線。棒グラフの挿入は、各遺伝子の曲線下面積(AUC)を示しています。
我々はさらに、異なる遺伝子セットに対するgRNAをデザインすることで、定義された細胞経路(プロテアソーム機能)以外でのCRISPRアルゴリズムの予測有用性を調べました。広く研究されている遺伝子10個(RNAi製品注文の過去の需要に基づく)をターゲットに、高スコアと低スコアのgRNAを10個ずつ選び、次世代シーケンサーで全indel形成についてテストしました(Figure 4)。高スコアのcrRNAの大部分(93%)(青色バー)は高効率の編集(40%以上のindel形成と定義)を示しましたが、低スコアのcrRNA(オレンジバー)のうち編集効率が高かったのは33%に過ぎず、全体的に機能性スコアが高いgRNAは、DNAレベルでのindel形成として測定される編集効率が高いことが示されました。
このようにして見つけたデザインルールを適用することで、目的のあらゆる遺伝子に対するカタログ試薬の作製が可能になり、また遺伝子ノックアウト能力が保証されたスクリーニング・ライブラリーの改良もできるようになりました。
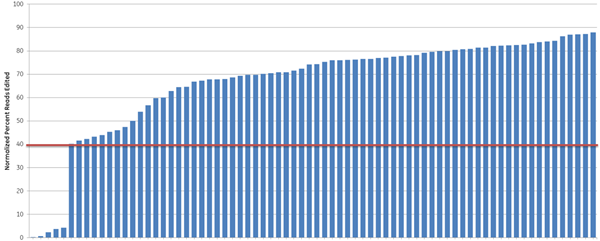

Figure 4. アルゴリズムスコアと編集効率との相関(indel形成のNGS解析)。10個の遺伝子を標的とするアルゴリズムスコアが最も高い10個のcrRNA(青色)とアルゴリズムスコアが最も低い10個のcrRNA(オレンジ色)を、Cas9を安定的に発現するHEK293T細胞にトランスフェクトし、次世代シーケンサーで編集を評価しました。Cas9-HEK293T細胞株を50 nM crRNA:tracrRNAでトランスフェクトしました。トランスフェクションの72時間後、細胞を溶解し、各crRNA部位にまたがるNexteraトランスポゾンアダプトアンプリコンを、トランスフェクトしていないサンプルからのマッチしたコントロールアンプリコンと同様に、処理したすべてのサンプルについて生成しました。サンプルはNextera 96-well index kitを用いてインデックスを作成し、MiSeq装置でのシーケンス用にプールした(ペアエンドリード、長さ2 x 300)。NGS quality filtering criteriaに合格したリードを参照ファイル(Bowtie2 v2.1.0)にアライメントしました。完全リードの割合を計算し、トランスフェクトしていないコントロールサンプルに対して正規化しました(Samtools v0.1.12a)。
Edit-Rアルゴリズムの検証
アルゴリズムが設計したgRNAがタンパク質レベルで効率的な機能遺伝子ノックアウトを予測する能力を調べるために、アルゴリズムのスコアと特定の機能アッセイにおける標的遺伝子のタンパク質レベルまたは活性への影響との相関性をテストしました。まず、TNFRSF1A(TNFRSF1Aタンパク質の可溶性部分をコード)を標的とする79のgRNAの機能性を、TNFRSF1A ELISAアッセイで測定されるTNFRSF1Aタンパク質レベルの減少を引き起こす能力について調べました。Figure 5Aは、細胞培地中のTNFRSF1Aタンパク質レベルに対するすべてのcrRNAの効果を、アルゴリズムが予測した機能性スコアで並べたものです。アルゴリズムスコアが高いcrRNAは、標的遺伝子タンパク質レベルの減少として測定される高い機能的遺伝子破壊を示しています。スコアが低いcrRNAの中では、機能性のばらつきが大きく、多くのガイドRNAが機能的遺伝子ノックアウトの能力が低いことを示しています(Figure 5A)。このことは、アルゴリズムスコアに基づき、低スコアから高スコアまで4つの四分位に分けられたcrRNAの機能性のボックスプロット表現によって明確に描かれています。中央値、下位四分位値と上位四分位値間のデータの分布、最小値と最大値は、アルゴリズムで設計された高スコアのcrRNAがタンパク質ノックアウトアッセイで機能性を高めていることを示しています(Figure 5B)。
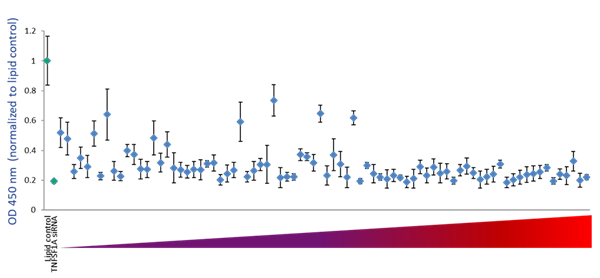

Figure 5. 可溶性TNFRSF1AのELISAアッセイによるTNFRSF1A crRNAの機能性の検討
A. Cas9(CAGプロモーター下)を組み込んだU2OS-プロテアソーム細胞を96ウェルプレートに10,000細胞/ウェルでプレーティングしました。プレーティングの24時間後、細胞を0.2 µg/ウェルのDF4を用いて50 nM crRNA:tracrRNAでトランスフェクトしました。TNFRSF1Aを標的とするsiRNAをポジティブコントロールとして用い、データはネガティブコントロール(脂質のみ)で正規化しました。トランスフェクションの72時間後に細胞培地を回収し、Quantikine Human sTNF RI/TNFRSF1A Quantikine ELISA Kit(R&D Systems)を用いて可溶性TNFRSF1Aをアッセイしました。
B. 溶性TNFRSF1AのELISAアッセイにおけるTNFRSF1A crRNAの機能性の箱ひげ図表示。crRNAは、低から高(Q1、Q2、Q3、Q4)までのアルゴリズムスコアに基づいて4つの四分位に分けられています。
更に、アルゴリズムが予測した機能性スコアと、下流の機能的効果による標的遺伝子のノックアウトとの相関を調べることで、Dharmacon Edit -Rアルゴリズムを評価しました。細胞生存に関与する3つの遺伝子、BCL2L1、PLK1、WEE1を標的とする可能性のある全てのcrRNAを、Cas9安定細胞にトランスフェクトし、アポトーシスアッセイを用いてアポトーシスを測定することによってテストしました。Figure 6Aは箱ひげ図データで、crRNAはアルゴリズム設計スコアに基づいて下半分(H1)と上半分(H2)の箱に分けられています。ここでも機能スコアの高いcrRNAは、スコアの低いデザインよりも強い表現型を示します。最後に、有糸分裂指数を用いて、リン酸化ヒストンH3(pSer10)陽性細胞の割合で測定した、有糸分裂制御に関与する遺伝子(PLK11とKIF11遺伝子ターゲット)のノックアウトの表現型効果とEdit-Rアルゴリズムスコアの相関を調べました(Figure 6B)。
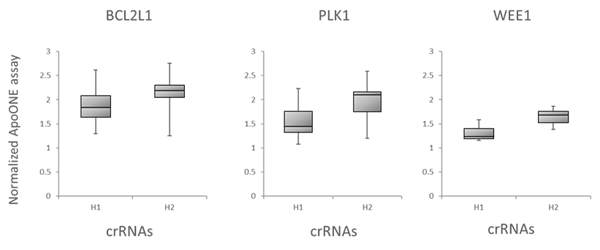
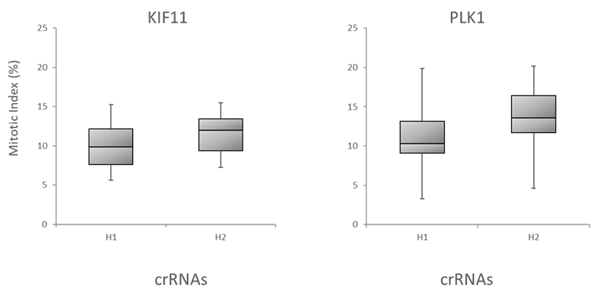
Figure 6. アルゴリズムスコアと機能アッセイの相関性
A. アポトーシスアッセイにおけるBCL2L1、PLK1またはWEE1を標的とするcrRNAの機能性の箱ひげ図。Cas9(CAGプロモーター下)を組み込んだU2OS-プロテアソーム細胞を96ウェルプレートに10,000細胞/ウェルでプレーティングしました。プレーティングから24時間後、細胞を0.2 µg/ウェルのDF4を用いて25nM crRNA:tracrRNAでトランスフェクトし、その48時間後にアポトーシスを解析しました。crRNAはアルゴリズム設計スコアに基づいて下半分(H1)と上半分(H2)に分けられました。
B. 有糸分裂指標アッセイにおけるKIF11またはPLK1を標的とするcrRNAの機能性の箱ひげ図。CAGプロモーター下にCas9を組み込んだU2OS-プロテアソーム細胞を96ウェルプレートに5,000細胞/ウェルでプレーティングしました。プレーティングから24時間後、細胞を0.1 µg/ウェルのDF4を用いて25nM crRNA:tracrRNAでトランスフェクトし、その48時間後に細胞を固定し、高含量分析によって分裂指数を分析しました。要約すると、細胞をPhospho-Histone H3 pSer10 AntibodyとGoat anti-Rabbit Secondary Antibodyで染色しました。核はHoechst色素で染色しました。有糸分裂指数は、有糸分裂中の細胞の割合(Phospho-Histone H3 シグナル陽性)で示しました。注:未処置のコントロールにおける有糸分裂指数は〜3%でした。データは箱ひげ図として示され、crRNAはEdit-Rの機能スコアに基づいて下半分(H1)と上半分(H2)に分けられています。
CRISPR-Cas9ガイドRNAの特異性に関する一考察
CRISPR-Cas9を用いて遺伝子をノックアウトするためのガイドRNAを選択する場合、gRNAの効率や機能が最も重要です。高性能のガイドRNA配列は、ゲノムDNAを切断し、改変されたmRNAから機能的タンパク質への翻訳をブロックする挿入と欠失の形成を通して、遺伝子を効果的に破壊しなければなりません。しかしながら、機能的なガイドRNAを決定することは、課題の半分でしかありません。ゲノムの不要な場所に永続的な変化を引き起こすことなく、最高のゲノム編集結果を得るためには、特異性の高いガイドRNAも必要です。
Cas9はガイドRNAの最初の20ヌクレオチドを使い、プロトスペーサー隣接モチーフ(protospacer-adjacent motif: PAM)(S. pyogenes Cas9ではNGG)の直上流にあるゲノムの配列と完全またはほぼ完全な相補性で塩基対を形成することで、ゲノムの位置を標的化します。複数の初期の研究で、まれに3つまでのミスマッチ(またはそれ以上)が許容され、オフターゲット認識や意図しないゲノム標的部位の切断につながることが実証されました(Fu, 2013; Hsu, 2013; Pattanayak, 2013)。一般的に、シード領域(PAMに最も近い~10塩基)のミスマッチは許容度が低く、隣接する2つのミスマッチはさらに許容度が低いです(Anderson, 2015)。しかし、ハプロイドヒトゲノムが30億以上の塩基から構成されていることを考えると、一つの遺伝子を標的とする複数のgRNAはともかく、一つのgRNAに対して起こりうるオフターゲッティングの結果を全て同定するのは大変な作業です。

Dharmacon オフターゲットアラインメントツールは、ミスマッチ塩基だけでなく、一塩基のギャップアラインメントも含め、各gRNAのオフターゲットの可能性のある全ての位置を検索します(Figure 1)。 機能と特異性の両方から効果的なgRNAをランク付けする場合、オフターゲッティング検索が不完全だとgRNAの選択が確実にできないため、これは重要です。
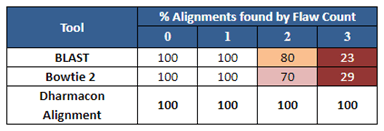
Figure 2は、ある特定の配列について、Dharmaconアライメントツールがいくつかの公開配列アライメントツールに対してどのように機能するかの例です。どのツールも1つの欠陥(欠陥:Flawとは1つのミスマッチまたはギャップ)を持つオフターゲット配列を同定していますが、2つか3つの欠陥については、公開ツールはゲノム中で起こりうるオフターゲッティングの結果を不完全に予測していることに注目してください。
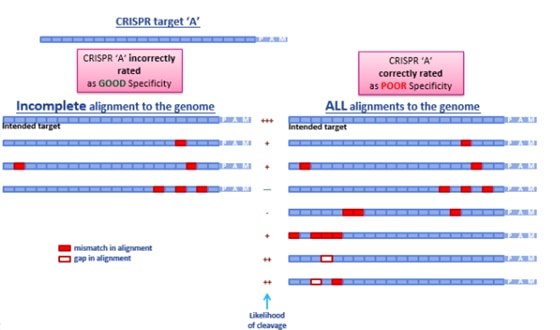
この不完全さが、オフターゲットの検索やガイドRNAのランキングにどのように反映されるかをFigure 3に示します。ギャップがあるオフターゲット候補のアラインメントが見つからないだけで、ガイドRNAは特異性が高いと誤って予測されます(左図)。これらのギャップアラインメントはオフターゲットの可能性が高く、この選択されたgRNAは特異性という点では低い候補となります。
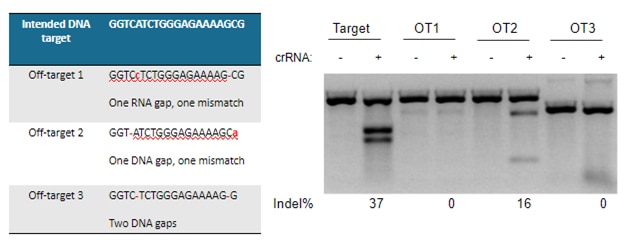
アライメントのずれによって引き起こされる実際のオフターゲットのデモンストレーションがFigure 4に示されており、予測されたすべてのオフターゲットgRNAが測定可能な効果につながるわけではないことも明確に示されています。
Edit-Rの独自のガイドRNA(化学合成crRNAおよびレンチウイルスsgRNA)設計アルゴリズムは、特異性解析結果を組み込んで、ヒト、マウス、ラットのモデル生物に対して、高い特異性とゲノム編集効率を持つ試薬を簡単に注文できるようにあらかじめ設計しています。遺伝子を検索し、利用可能なデザインから選択するだけで、ご利用いただけます。
Order products
Cas9ヌクレアーゼ
細胞タイプに最適なプロモーターを選択してCas9発現細胞を得るか、DNAフリーのオプションを選択できます。
CRISPRコントロールと検出プライマー
CRISPRコントロールと検出プライマー
CRISPRガイドRNA
Cas9切断をガイドする、高品質ですぐに使用できるレンチウイルスおよび化学合成試薬
CRISPR-Cas9ゲノム編集
信頼性の高いゲノムエンジニアリング用に最適化されたツール
プール化sgRNAまたはアレイ化crRNA
ハイスループットでのゲノム編集研究のためのプール化sgRNAまたはアレイ化crRNAライブラリー
Helpful resources
化学合成RNAを使用したCRISPR-Cas9ゲノム編集:From Start To Finish
Read app note
CRISPR-Cas9ゲノム編集ワークフロー:Edit-R Cas9と化学合成crRNAおよびtracrRNAを使用した機能的ノックアウトの実施
Watch video
Edit-R CRISPR-Cas9ゲノム編集プラットフォーム