

Knockout your target gene
Find All-in-one lentiviral sgRNA for guaranteed editing of any human or mouse gene
| All-in-oneのメリット: | 理想的な用途: |
|---|---|
|
|
|
|
|
|
|
|
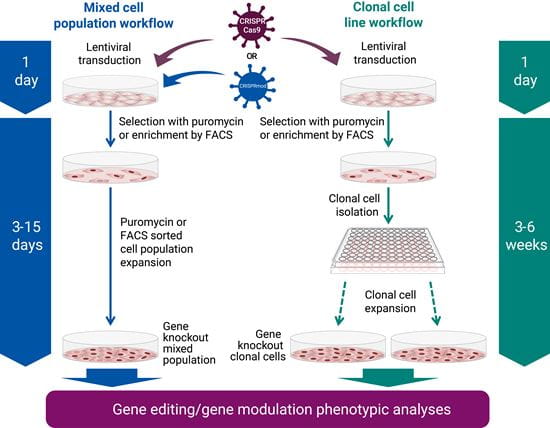
遺伝子導入は、試薬を解凍し、適量を細胞に加えるだけ。その後、ピューロマイシンを用いて目的の細胞を選択するか、FACSを用いて濃縮することが簡単にできます。
ピューロマイシンセレクションを使用する場合、トランスダクション後24~48時間で増殖培地をピューロマイシンセレクション培地に置き換えます。その後、選択培地で正常に増殖するまで、細胞を毎日観察します。成長が安定したら、目的とする細胞集団の単離および/または細胞増殖を行うことができます。
また、EGFP蛍光レポーターオプションを使用すると、導入後72時間でFACSセレクションにより編集細胞を濃縮することが可能です。FACSによる濃縮ワークフローは、初代細胞のような短命の細胞タイプに特に有効です。
CRISPRaおよびCRISPRiについては、混合細胞集団のワークフローを推奨します。
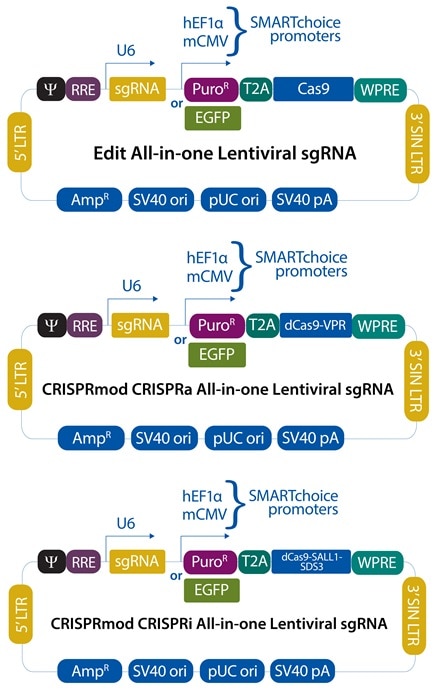
CRISPR sgRNA + Cas9の発現を1つのベクターに組み合わせ、ゲノム編集を容易にします。
ヒトゲノム全体をカバーする高力価プール化スクリーニングライブラリー
CRISPR sgRNA + dCas9-VPRの発現を1つのベクターに組み合わせ、遺伝子転写活性化を容易にします。
ヒトゲノム全体をカバーする高力価のCRISPRaプール化スクリーニングライブラリー
CRISPR sgRNA + dCas9- SALL1-SDS3の発現を1つのベクターに組み合わせ、遺伝子転写抑制を容易にします。
ヒトゲノム全体をカバーする高力価のCRISPRiプール化スクリーニングライブラリー
DharmaconTM All-in-oneシステムAll-in-oneシステムは、単一試薬、より簡単なデリバリー、選択しやすさという、最もシンプルなCRISPRワークフローを提供します。 単一試薬1つのベクターでCRISPRヌクレアーゼと遺伝子特異的ガイドRNAを発現させることにより、導入と選択を連続して行う必要がありません。 より簡単なデリバリーレンチウイルスのパッケージングにより、ほぼすべての細胞タイプに簡単に導入することができます。毒性のあるトランスフェクション試薬やエレクトロポレーション技術を必要としないため、All-in-oneシステムは、トランスフェクションが困難な細胞や初代細胞におけるゲノム編集や転写調節に特に有用です。 また、transduction-ready試薬は、クローニングやin vitro転写のステップを必要としません。 選択しやすさピューロマイシン耐性遺伝子またはEGFPマーカーを選択することで、ベクターの導入に成功した細胞を容易に選択することができます。EGFPは、蛍光が発現したらすぐにFACS解析を行うことができるため、標的細胞の迅速な濃縮に推奨されます。
プロモーターオプションプロモーターの選択により、お客様の細胞株で最も活性の高いプロモーターを選択することができます。 |
 |
Efficient CRISPRmod CRISPRa All-in-one Puro-dCas9-VPRによる効率的な転写活性化
HEK293TまたはHCT-116細胞を96ウェルプレートにそれぞれ20,000または5,000細胞/ウェルでプレーティングし、POU5F1、TTN、IL1R2を標的とするsgRNAを含むCRISPRa All-in-oneレンチウイルスまたはnon-targetingコントロールレンチウイルスを0.3 MOIで形質導入しました。細胞は2.5 µg/µLのピューロマイシンで5日間選択し、増殖させ、導入後2週間目に採取しました。RT-qPCRを用いて相対的な遺伝子発現を算出しました。各遺伝子の相対発現は、ACTBをハウスキーピング遺伝子として∆∆Cq法で算出し、non-targetingコントロールに対して正規化しました。
感染多重度(MOI: multiplicity of infection)を増加することにより標的活性化が向上する
HEK293T細胞を96ウェルプレートに20,000細胞/ウェルでプレーティングし、POU5F1、TTNを標的とするsgRNAを含むCRISPRa All-in-oneレンチウイルスまたはnon-targetingコントロールレンチウイルスを図に表示されたMOIで形質導入しました。細胞は2.5 µg/µLのピューロマイシンで5日間選択培養し、増殖させ、形質導入後1週間または2週間で採取しました。RT-qPCRを用いて遺伝子の相対的発現量を算出しました。各遺伝子の相対発現は、ACTBをハウスキーピング遺伝子として∆∆Cq法で算出し、non-targetingコントロールに対して正規化しました。
ソーティングされていないU2OS細胞におけるOCT4タンパク質発現の誘導
U2OS細胞を96ウェルプレートに10,000細胞/ウェルでプレーティングし、POU5F1を標的とするsgRNAを含むEGFP CRISPRa All-in-oneウイルスまたはnon-targetingコントロールレンチウイルス(NTC2)を0.5 MOIで形質導入しました。細胞は継代し、96ウェルプレートおよび6ウェルプレートに展開しました。導入後1週間経過した96ウェルプレートで培養した細胞からRNAを抽出しました。RT-qPCRを用いて、遺伝子の相対発現量を算出しました。各遺伝子の相対発現は、ACTBをハウスキーピング遺伝子として∆∆Cq法で算出し、non-targetingコントロールに対して正規化しました。
EGFP mCMV CRISPRa All-in-oneウイルス粒子によるソーティングされていないU2OS細胞における OCT4タンパク質の活性化
U2OS細胞を96ウェルプレートに10,000細胞/ウェルでプレーティングし、POU5F1を標的とするsgRNAを含むEGFP CRISPRa All-in-oneウイルスまたはnon-targetingコントロールレンチウイルス(NTC2)を0.5 MOIで形質導入しました。 |
CRISPRmodによる効率的な遺伝子転写抑制 CRISPRi All-in-oneシステム
EK293T、U2OS、またはHCT-116細胞を96ウェルプレートに20,000、10,000、または5,000細胞/ウェルでプレーティングし、PPIB、SEL1Lを標的とするsgRNAを含むCRISPRi All-in-oneレンチウイルスまたはnon-targetingコントロールレンチウイルスを0.3 MOIで形質導入しました。細胞は2.5 µg/µLのピューロマイシンで5日間選択し、増殖させ、形質導入後の指定された時点で採取しました。RT-qPCRを用いて、遺伝子の相対発現量を算出しました。各遺伝子の相対発現は、ACTBをハウスキーピング遺伝子として∆∆Cq法で算出し、non-targetingコントロールに対して正規化しました。
ピューロマイシン選択により、遺伝子転写抑制が改善される
U2OS細胞を96ウェルプレートに10,000細胞/ウェルでプレーティングし、PPIB、SEL1L、TFRCを標的とするsgRNAを含むCRISPRi All-in-oneレンチウイルスまたはnon-targetingコントロールレンチウイルスを0.3 MOIで形質導入しました。細胞は2.5 µg/µLピューロマイシン(該当する場合)で5日間選択し、増殖させ、形質導入後1週間または2週間後に採取しました。RT-qPCRを用いて、遺伝子の相対発現量を算出しました。各遺伝子の相対発現は、ACTBをハウスキーピング遺伝子として∆∆Cq法で算出し、non-targetingコントロールに対して正規化しました。
EGFP陽性のJurkat細胞における遺伝子転写抑制の増加
Jurkat 細胞を 25,000細胞/ウェルでプレーティングし、CD46 を標的とする sgRNA を含むCRISPRi All-in-oneウイルス(CRISPRi)またはnon-targetingコントロールレンチウイルス(NTC2)を0.3 MOIで形質導入しました。細胞は2つに分け、一方は1週間培養し、mRNA抽出のために採取しました(形質導入1週間後、Pre-FACS)、もう一方は3週間培養し、PBSで洗浄後、FACSバッファ(10 mM Tris-HCl - pH 7.5, 25 mM HEPES, 1% FBS含有1X PBS)中に再懸濁させました。EGFP陽性細胞をFACSでソートし、1ウェルあたり〜50,000個のEGFP陽性細胞を含むソーティングを24ウェルプレートに入れました。細胞は1週間増殖させ、導入後4週間目にRNAを採取しました(Post-FACS)。RT-qPCRを用いて、遺伝子の相対発現量を算出しました。各遺伝子の相対発現は、ACTBをハウスキーピング遺伝子として∆∆Cq法で算出し、non-targetingコントロールに対して正規化しました。 |
プール化CRISPRスクリーニングライブラリーは、ゲノムレベルの細胞システム解析に不可欠であり、薬剤や病原体のメカニズム、遺伝子の必須性など、数多くの生物学的な疑問の解決に役立ちます。しかし、レンチウイルスCRISPR構コンポーネントを順次導入することは、特にトランスフェクションが困難な細胞や初代細胞では困難です。さらに、Cas9 エフェクターと遺伝子特異的シングルガイドRNA(sgRNA)の両方を高力価レンチウイルスライブラリーに発現するパッケージングベクターは、特にCRISPR活性化(CRISPRa)またはCRISPR干渉(CRISPRi)に必要な大型の不活性化Cas9(dCas9)融合体では、労力がかかります。
当社の高力価で均一に分布されたDharmacon™ all-in-one レンチウイルスプール化CRISPRライブラリーは、これらの課題を克服します。
厳密なレンチウイルスプール化ライブラリー生産から始まる高品質なプール化スクリーニング
Dharmaconの3種類のall-in-oneレンチウイルスプール化ライブラリーは、いずれもインプットsgRNAの回収率が98%以上と高く、均一に分布していることが次世代シーケンサー(NGS)で検証したところ示されています。

Dharmacon™ all-in-oneレンチウイルスプール化ライブラリーによる堅牢な機能的力価
all-in-oneプール化ライブラリーとライブラリーに一致するデリバリーコントロールの力価検証を2つの方法で行いました。すべてのプールライブラリーは機能的に力価が検証されています。2つの方法を比較すると、p24の力価検証は実際の値を過大評価していることがわかります。
