Freedom with custom pooled library designs for your experimental needs. Your custom library can have:
- Pools of lentiviral sgRNA or shRNA
- Customizable promoter and reporter options, including inducible promoters
- Complete workflow planning tools and protocols
- Human and mouse options available
Can't find a catalog screening library to meet your needs?
We can help you design your own custom lentiviral pools. Select from our range of algorithm-optimized reagents to develop the ideal screening resource for CRISPR-Cas9 knockout or RNAi knockdown screens.
Custom lentiviral pooled screening libraries offer an efficient method for screening large numbers of lentiviral sgRNAs or shRNAs without the requirement of automation. All you need to do is select the product format, the number of genes, constructs per gene, and species.
Whether you need to perform a CRISPR knockout screen with lentiviral sgRNAs or a knockdown RNAi screen with shRNA a custom library can be built to your experimental requirements.
Ensure reproducibility and accurate hit identification
Library construction, pooling techniques, and high-throughput sequencing compatible screening workflows have all been experimentally validated. Pools are provided as concentrated lentiviral particles for transduction into dividing and non-dividing cells.
Experimentally validated protocols and calculation sheets are readily available to help you with every aspect of the experiment from determining relative titer in your specific cells to calculating exactly how much volume you will require to complete the experiment. Our Bioinformatic analysis protocol provides full instructions on using open source software to run the analysis from the NGS data.
To learn more about the critical parameters of successful pooled lentiviral screening, including the conditions necessary for maintaining a high fold-representation, please download the following publication: Ž. Strezoska, A. Licon, Optimized PCR Conditions and Increased shRNA Fold Representation Improve Reproducibility of Pooled shRNA Screens. PLoS One 7, e42341 (2012).
Specifications
Titer: Constitutive vectors, 5 x 108 TU/mL (± 20%); inducible vectors, 1 x 107 TU/mL (± 20%); all products are purified lentiviral particles with functional titers determined by qPCR. Pool size: minimum of 50 total constructs up to 12,000 constructs per pool Volume: Minimum of 100 uL up to 5 mL per pool; delivered in 25 µL aliquots
Custom lentiviral pooled libraries can be generated from any of the following product lines
Edit-R Predesigned Lentiviral sgRNA
Guaranteed to edit your target! Algorithm-optimized sgRNA for genome-wide coverage of human or mouse genes. Provided as high-titer lentiviral particles and glycerol stocks.
| Number of targeted genes (protein-coding only) |
Available controls* | Compatible NGS Analysis kit | |
|---|---|---|---|
| Human | 19,000 | 100 NTC 340 positive | Dharmacon NGS Library Prep Kits |
| Mouse | 19,680 | 100 NTC 340 positive | Edit-R Mouse Pooled sgRNA Indexing PCR and Sequencing Primer Kits |
Edit-R predesigned All-in-one lentiviral sgRNA , CRISPRmod CRISPRa All-in-one Lentiviral sgRNA, and CRISPRmod CRISPRi All-in-one Lentiviral sgRNA
CRISPR workflows simplified with Dharmacon™ All-in-one vectors. The All-in-one platform combines the CRISPR nuclease or effector and single guide RNA into a single lentiviral packaged vector – for the simplest gene knockout/modulation workflow available.
| Number of targeted genes (protein-coding only) |
Available controls* | Compatible NGS Analysis kit | |
|---|---|---|---|
| Human | 19,000 | 100 NTC 340 positive | Dharmacon NGS Library Prep Kits |
| Mouse* | 19,680 | 100 NTC 340 positive | Edit-R Mouse Pooled sgRNA Indexing PCR and Sequencing Primer Kits |
SMARTvector Lentiviral shRNA and SMARTvector Inducible Lentiviral shRNA
Advanced rationally-designed microRNA-based shRNA for human, mouse, and rat genes using our patented scaffold and design characteristics. Choose from multiple promoter and reporter options for ultimate experimental flexibility.
| Number of targeted genes (includes lncRNA genes) | Average designs per gene | Available controls* | Compatible NGS Analysis kit | |
|---|---|---|---|---|
| Human | 19241 | 9.5 | 100 NTC 272 positive | SMARTvector Indexing PCR and Sequencing Primer Kits |
| Mouse | 21745 | 9.2 | 100 NTC 272 positive | SMARTvector Indexing PCR and Sequencing Primer Kits |
| Rat | 22646 | 9.2 | 100 NTC 272 positive | SMARTvector Indexing PCR and Sequencing Primer Kits |
shMIMIC Lentiviral microRNA and shMIMIC Inducible Lentiviral microRNA
Innovative microRNA vector design coupled with your choice of optimal promoter and reporter for over-expression of mature microRNAs.
| shMIMIC Lentiviral microRNA | Number of mature microRNAs | Number of unique designs | Available Controls* | Compatible NGS Analysis kit |
|---|---|---|---|---|
| Human | 2580 | 2555 | 30 NTC 80 positive | SMARTvector Indexing PCR and Sequencing Primer Kits |
| Mouse | 1913 | 1896 | 30 NTC 80 positive SMARTvector | Indexing PCR and Sequencing Primer Kits |
GIPZ Lentiviral shRNA (Decode)
Efficient gene silencing with a microRNA-adapted shRNA design. Available as lentiviral vector constructs or high-titer lentiviral particles for human and mouse
| Number of targeted genes | Average designs per gene | Available controls* | Compatible NGS Analysis kit | |
|---|---|---|---|---|
| Human | 18205 | 5.3 | 1 NTC 2 positive | Decode Indexing PCR and Sequencing Primer Kit |
| Mouse | 18045 | 5.6 | 1 NTC 2 positive | Decode Indexing PCR and Sequencing Primer Kit |
-
SMARTvector Lentiviral shRNA Pooled Libraries
Constitutive shRNA expression for optimized functional analysis screens -
SMARTvector Inducible Lentiviral shRNA Pooled Libraries
Inducible shRNA expression for regulatable and controlled functional analysis screens -
shMIMIC Lentiviral microRNA Pooled Libraries
Perform functional screening of hundreds or thousands of microRNAs without high-throughput automation -
shMIMIC Inducible Lentiviral microRNA Pooled Libraries
Tight control of microRNA expression for powerful functional screening of hundreds or thousands of microRNAs. -
SMARTvector Indexing PCR and Sequencing Primer Kit
For use with SMARTvector and shMIMIC pooled libraries on Illumina systems
High quality pooled screening begins with rigorous library production
All lentiviral library pools are created using experimentally validated methods that ensure uniform representation of constructs in every lentiviral pool. After pooling, DNA is prepared from E. coli cultures and analyzed by high-throughput sequencing to evaluate construct representation and identity. This quality control allows us to verify that > 95% of constructs are recovered after the pooling process and the abundance of 70% of the constructs is less than 5-fold different from each other and the abundance of 90% of the constructs is less than 25-fold different from each other. This quality control provides confidence in the uniformity of our pooled screening libraries and the ability to detect changes in construct representation
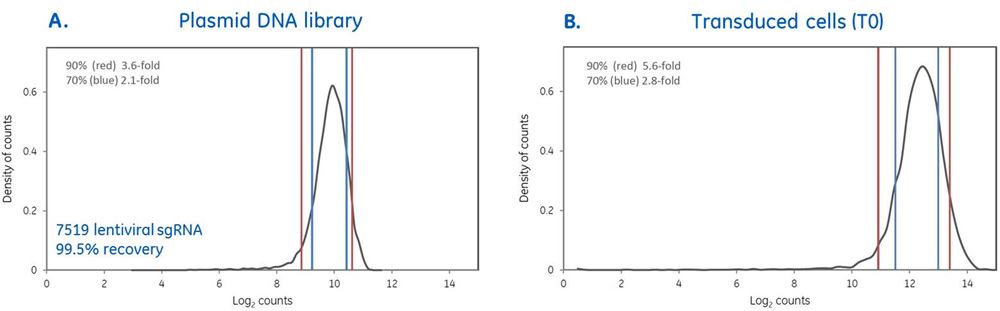
High quality pooled screening begins with rigorous lentiviral pooled library production. In this example a pooled library comprised of 7519 lentiviral sgRNAs targeting human kinases was produced and the quality of the plasmid DNA library was verified by next-generation sequencing (NGS). Counts per million mapped reads were obtained to determine percent-recov1ery of input sgRNAs (99.5%) and the distribution of 90% and 70% of the sgRNAs in the pool are within 3.6- and 2.1- fold of each other, respectively (Panel A.). The pooled plasmid DNA library was then packaged into lentiviral particles and transduced at MOI 0.3 into U2OS cells from which genomic DNA was isolated at 24 hours post-transduction (T0), PCR-amplified and prepped for NGS. Distribution of lentiviral sgRNAs was not substantially affected, indicating rigorous production and maximized retention of constructs from production into pooled screening (Panel B.).
Successful pooled screening requires a high construct fold representation
Critical to the success of your screen and identification of quality hits is performing the screen at a high fold representation (the extent to which any given construct (e.g., shRNA) in a pooled library will be represented in the screen). High construct representation results in a greater degree of reproducibility between biological replicates and ensures that there is a sufficient experimental window for detection of changes in representation after phenotypic selection.
Horizon provides the tools necessary to reproducibly identify hits with confidence:
- All constitutive promoter lentiviral screening libraries are provided as concentrated (≥ 108 TU/mL) lentiviral particles in sufficient quantity to transduce multiple biological replicates. Libraries with an inducible reporter are provided at ≥ 107 TU/mL.
- Optimized and experimentally validated product-specific PCR primers are designed to efficiently amplify genomic DNA with minimal bias and allow downstream high-throughput sequencing analysis of construct abundance
- Sufficient quantity of lentiviral particles, PCR and sequencing primers allow the maintenance of high fold representation throughout the entire screening process
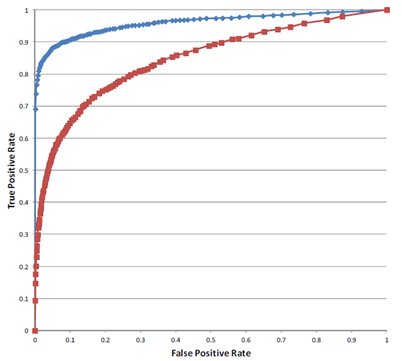
The Receiver Operating Characteristic (ROC) curves for a screen of 10,000 shRNAs at 100- (red) and 500-fold (blue) shRNA representation for detecting two-fold enrichment and depletion. The 500-fold screen has a larger area under the ROC curve and a higher true positive rate than the 100-fold screen, demonstrating the superior ability of the 500-fold screen to detect enriched and depleted shRNAs.
Reproducibility between replicates is maintained through PCR steps by following experimental protocols
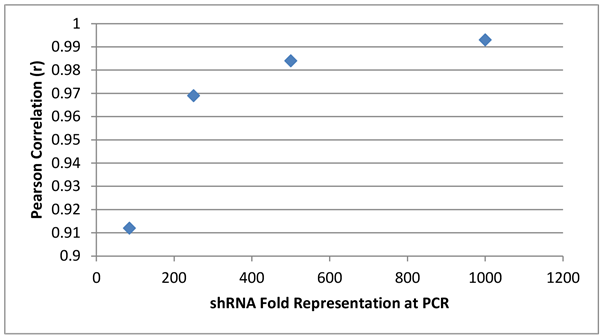
The Decode phosphatase library was transduced into HEK293T cells at 1,000-fold representation. Cells were selected with puromycin for 72 hours and genomic (gDNA) was isolated. Decode PCR primers were used to amplify shRNA sequences at indicated fold representations. Technical replicates of each PCR were compared to determine reproducibility (Pearson Correlation), where 1.0 indicates that the samples are 100% identical.
Successful pooled screening requires a high fold representation of each construct
Each step of the pooled screening workflow, from transduction to hit identification, has been empirically tested. Amplification conditions were identified to ensure uniform amplification and high reproducibility (Strezoska et al. 2012). Horizon pooled screening systems includes Illumina-adapted PCR primers for identification of hairpin sequences from gDNA by high-throughput sequencing on Illumina instrumentation. Vector-specific primer pairs are optimized for efficient amplification of the construct while minimizing thermodynamic bias and variation in representation. In addition, PCR primers have built-in adaptor and index sequences that allow the researcher to easily move from PCR amplification to Illumina high-throughput sequencing. Direct identification of hairpin insert facilitates data analysis and ensures accurate target gene identification.
PCR amplification and Illumina high-throughput sequencing workflow
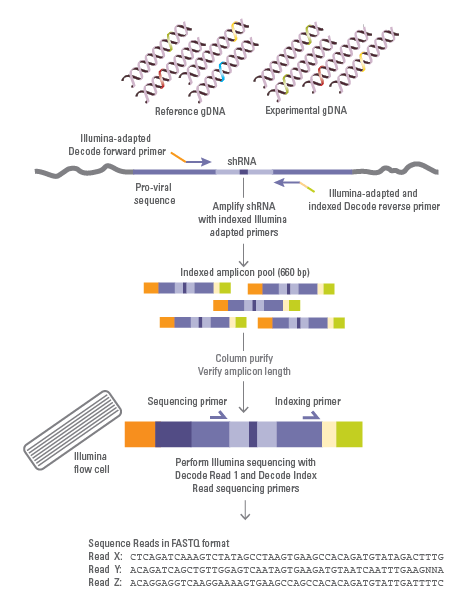
Illumina-adapted primers and Phusion Hot Start II High-Fidelity DNA Polymerase are used to PCR amplify integrated shRNA and sgRNA sequences and add Illumina flow-cell binding sequences. (note: formats have product-specific kits) The resulting amplicons are run on Illumina platform sequencers, using the sequencing primers provided. Constructs that are enriched or depleted during the screen are identified as hits, and the genes that they target are identified. Hits can be confirmed and studied further using individual sgRNA or shRNA constructs.
Next-generation sequencing protocol is reproducible so that experimental noise is reduced and hits are more easily identified
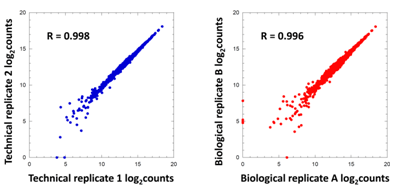
The Decode Phosphatase pooled screening library was transduced into HEK293T cells at 1000-fold shRNA representation. Cells were selected with puromycin for 72 hours and genomic DNA (gDNA) was isolated. Decode PCR and Sequencing primers were used to amplify and sequence gDNA samples as two technical duplicates (blue) and two different transductions (red) and counts analyzed by Illumina high-throughput sequencing. The log2 counts of shRNAs for each of the replicates are plotted against each other.