Strict R Inducible Cas9レンチウイルスシステム
精密な遺伝子ノックアウトを可能にする二重誘導制御:仕組み
System OFF:誘導剤非存在下での厳密な制御
ドキシサイクリンおよびShield1が存在しない場合、本システムは不活性状態を維持します。TRE3Gプロモーターからの最小限の転写リークによりデグロンタグ付きCas9が産生される場合でも、当該Cas9は速やかにプロテアソームによって分解されるため、バックグラウンドのゲノム編集活性は最小限に抑えられます。
System ON:制御された高効率ゲノム編集
ドキシサイクリンを添加すると、TRE3Gプロモーターからの強力な転写が誘導されます。さらにShield1を同時に添加することで、デグロンタグ付きCas9タンパク質が安定化され、標的特異的sgRNAの存在下で高効率かつ精密な遺伝子ノックアウトが可能になります。
この二重レベルの制御により、遺伝子編集実験において厳密な時間制御が実現されます。
Highlights
- 低分子誘導によるCas9発現を用いた、遺伝子編集の厳密かつ調整可能な制御
- Cas9ヌクレアーゼによる、高効率かつ高忠実度なDNA切断と、最小限のオフターゲット効果
- ドキシサイクリンおよびShield1の添加によりCas9の誘導発現および安定化を可能にするTet デグロンシステムの統合
- 単一のレンチウイルスベクターによるデリバリーで、既存の実験ワークフローへの容易な組み込みが可能
- 機能喪失研究、機能ゲノミクス、スクリーニング用途に適した、高品質かつ精製済みレンチウイルス粒子(≥1×10⁷TU/mL)を提供し、細胞毒性を最小限に抑制
- 実験設計に応じて選択可能な、ハイグロマイシン耐性、ブラスティシジン耐性、またはEGFP蛍光レポーターオプション
Strict R Inducible Cas9レンチウイルスベクターの模式図
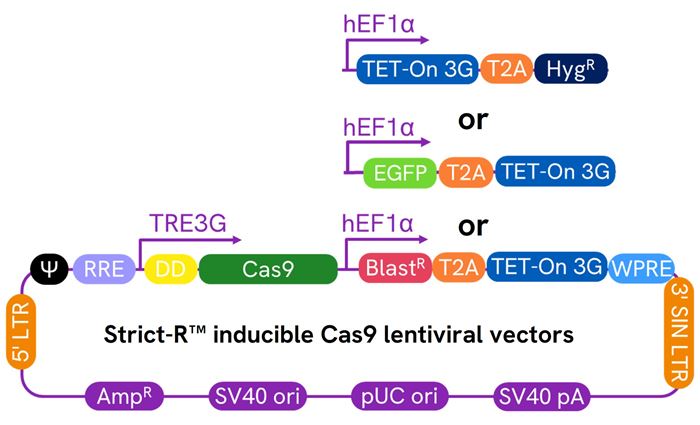
Strict R™ Inducible Cas9レンチウイルスシステムによる転写および翻訳後制御

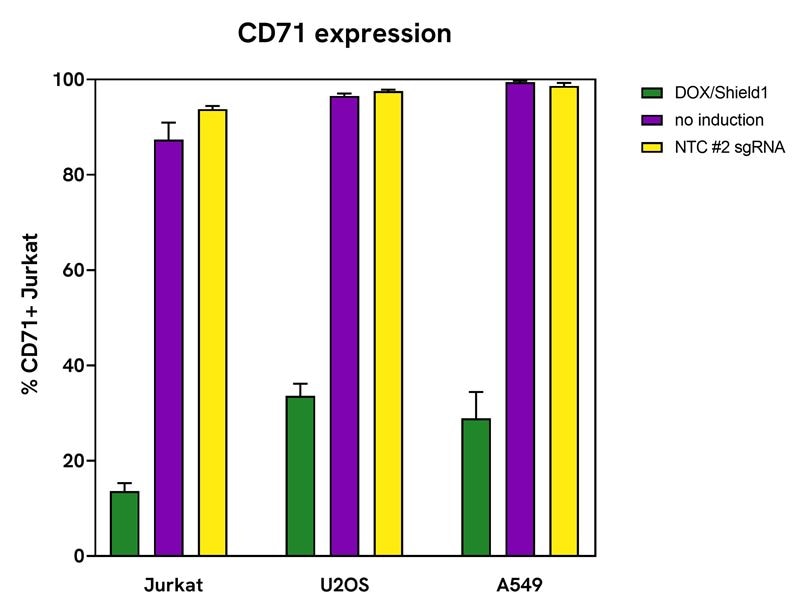
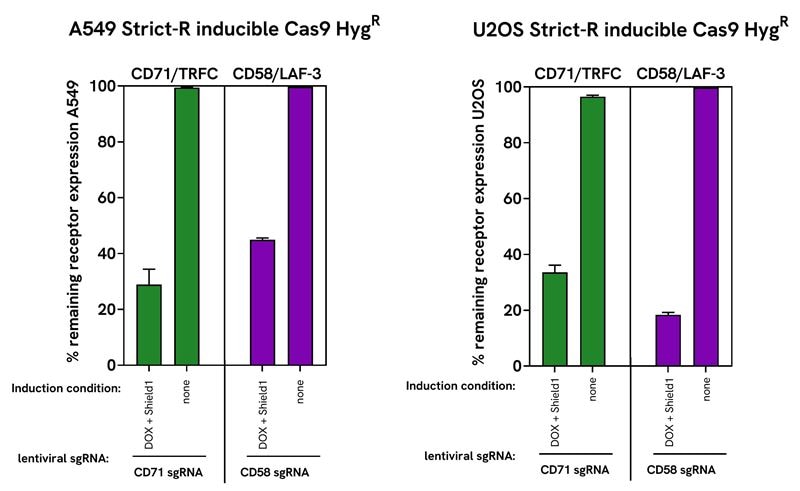
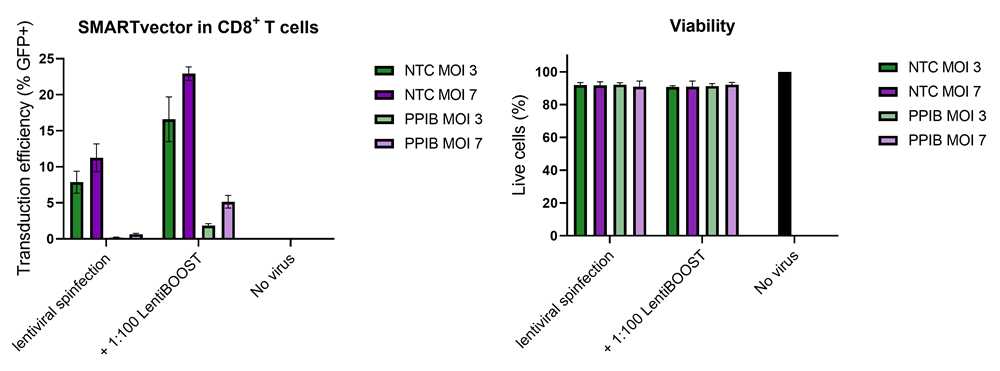
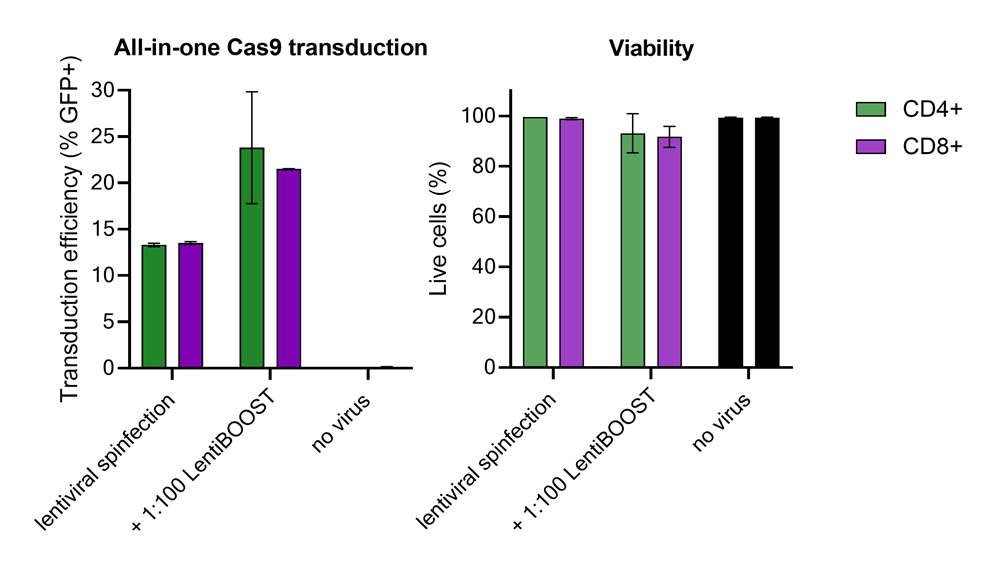
Dharmacon™ Strict R™ Inducible Cas9 レンチウイルスシステムの模式図を示しています。ドキシサイクリンおよびShield1非存在下では、本システムは「OFF」状態となります。TRE3Gプロモーターからのリーキーな転写により、FKBP12由来の不安定化ドメイン(デグロン)と融合したCas9が翻訳されますが、当該タンパク質は速やかにプロテアソーム分解を受けます。ドキシサイクリンの添加によりTRE3Gプロモーターからの強力な転写が誘導され、さらにShield1の添加によってCas9タンパク質が安定化されます。これにより、遺伝子特異的sgRNAの存在下で、強力な標的遺伝子編集およびノックアウトが可能になります。本図はBioRender.comを用いて作成されています。