Edit-R predesigned lentiviral sgRNA
Single guide RNA expressing vectors for effective and accurate gene knockout
- Guaranteed to edit the target gene of interest
- Transduction-ready RNAs eliminate cloning and in vitro transcription steps
- Algorithm-optimized to maximize the likelihood of functional protein knockout and reduce off-target editing
- Available as glycerol stocks and high-titer purified particles
Edit-R predesigned lentiviral sgRNA
1Start Here
Functional and specific targeting for high-confidence gene knockout results
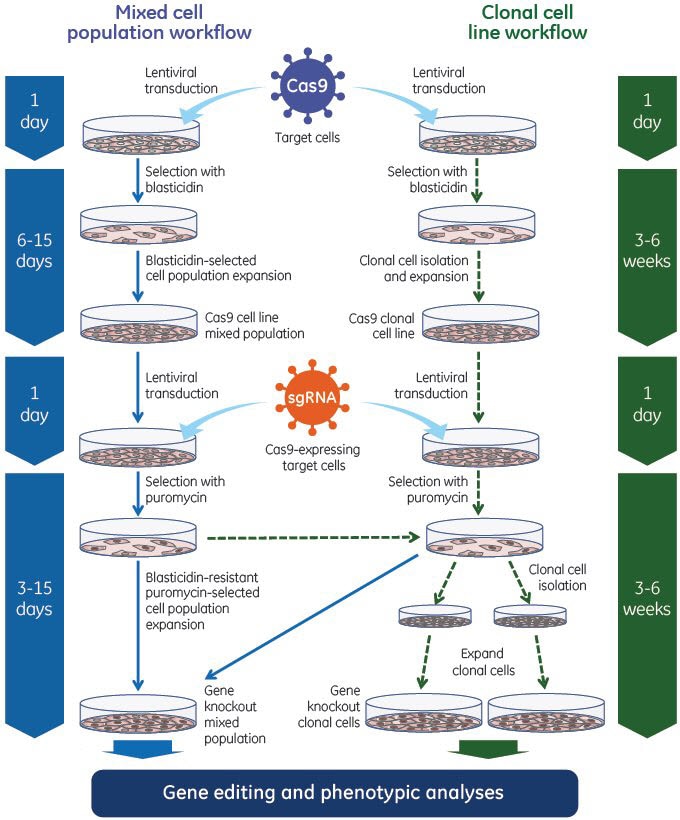
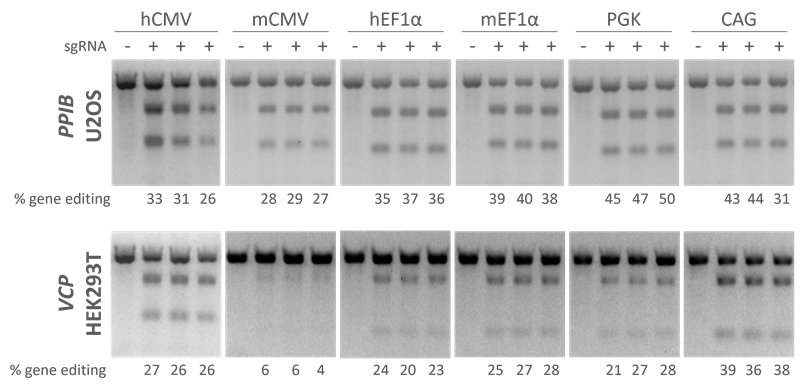
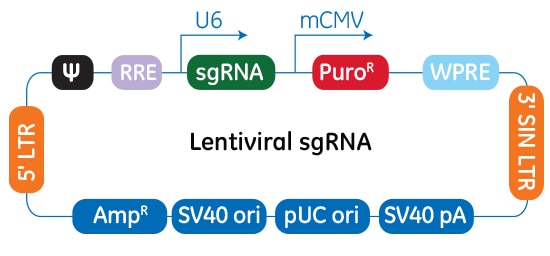
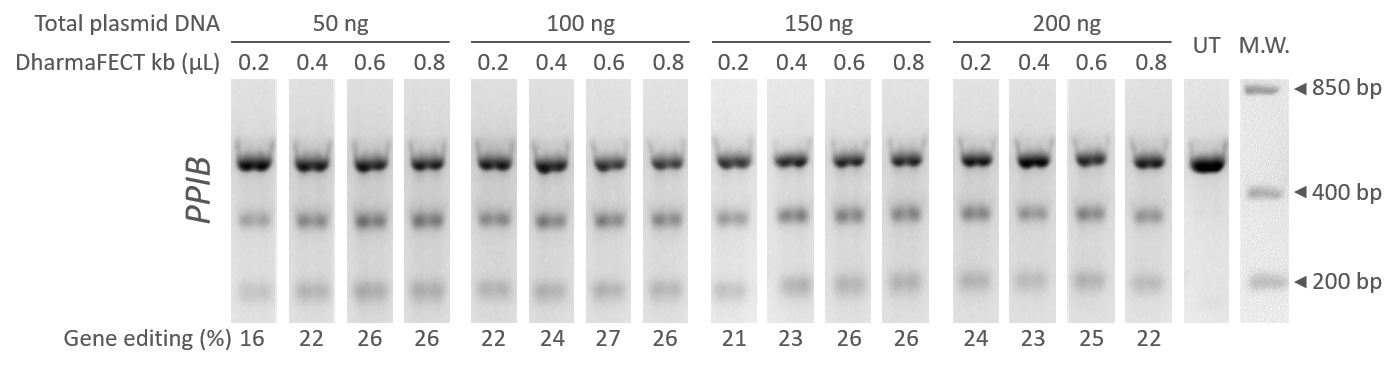
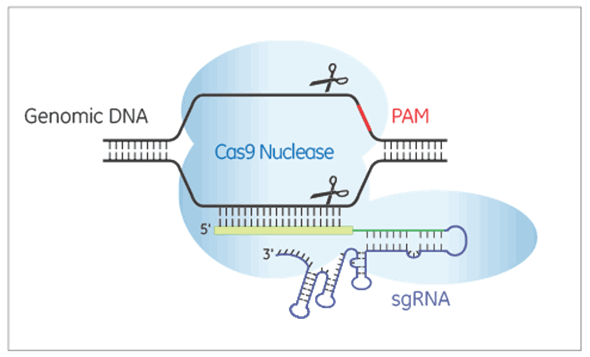
Edit-R Lentiviral sgRNAs express RNA for guiding Cas9 nuclease to create double-strand breaks in the target DNA. In the Edit-R lentiviral sgRNA vector backbone, the gene-specific guide RNA is expressed under the control of a human U6 promoter, while expression of the puromycin resistance marker (PuroR) is driven from the mouse CMV promoter and allows for rapid selection of cells with integrated sgRNA.
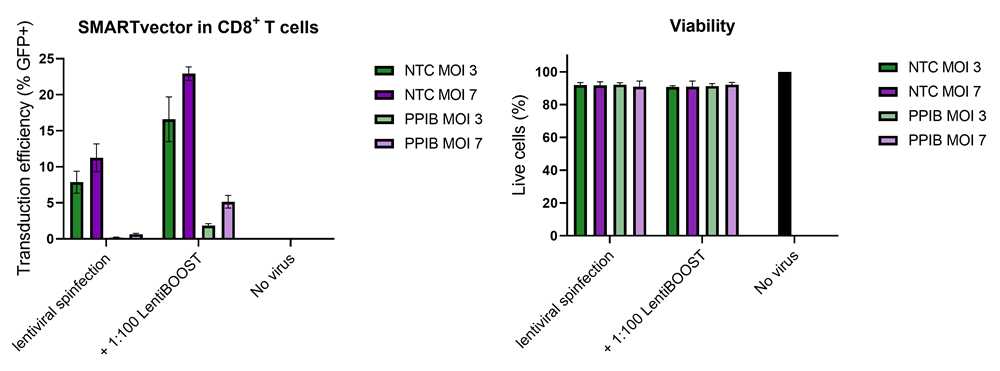
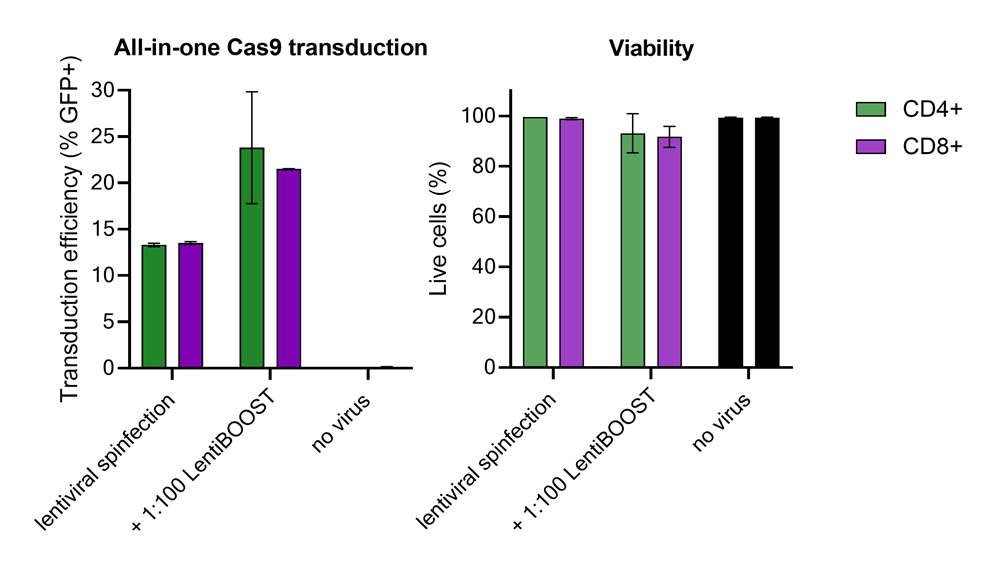
Each Edit-R Lentiviral sgRNA is specific to the gene and genomic site of interest. Generating knockouts in difficult-to-transfect cells or following up hits from a pooled lentiviral sgRNA library screen are key applications of this guide format.
While CRISPR-Cas9 is a highly effective tool for interrogating gene function, not all guide RNAs are effective in attaining functional protein knockout. To address this problem, Horizon developed an algorithm that is trained to select guides that give the highest likelihood of generating a functional gene knockout, not just creating an insertion or deletion.
New! Edit-R human sgRNA designs have been updated to the latest RefSeq in 2025 providing the most specific and genomically relevant guides for producing efficient protein knockout. This allows the Edit-R algorithm to target the latest genome annotations more accurately and efficiently providing you with the best solution for your research needs. Please reach out to Scientific Support if you have any questions or read our blog on revvity.com.
All guide RNA designs are top algorithm picks for each gene; qualitative ranks for functionality and specificity allow you to fine tune guide RNA choice to your specific application. The functionality score is a predicted indication of how likely this guide is to produce a functional knockout. The specificity score is based on the predicted risk of cutting activity at potential off-target sites. To learn more, visit our algorithm for Edit-R guide RNA page.